
앞 편에 이어, H(저산소) 트리거와 네 트리거가 서로 얽히는 방식을 살펴봅니다. 인용된 참고문헌은 원고 그대로입니다.
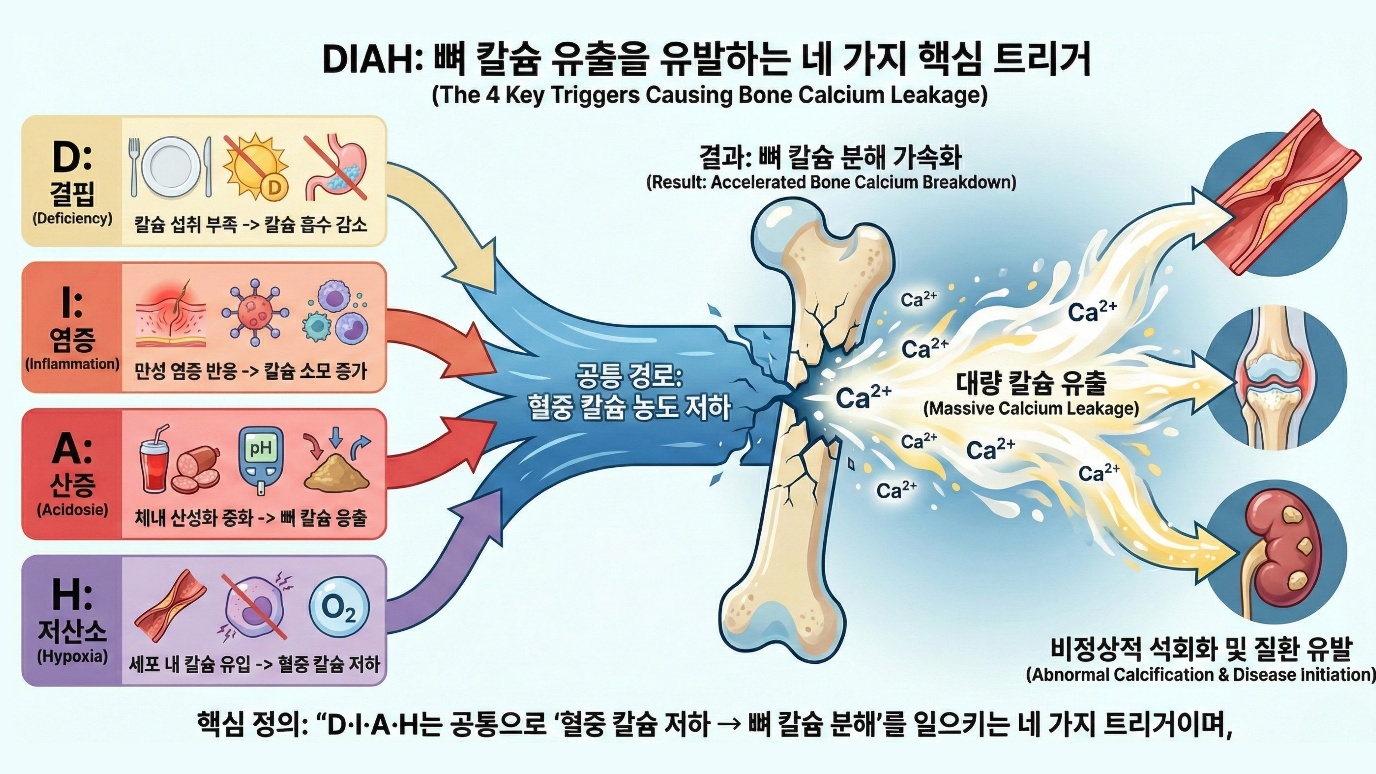
4. H 트리거: 저산소(Hypoxia) - 산소 부족에 칼슘이 세포로 과잉 유입된다
산소는 생명의 근본입니다. 유기호흡을 하는 모든 생명체는 산소가 필요합니다. 미토콘드리아에서 산소가 전자전달계의 최종 전자 수용체가 됩니다. 산소 없이는 ATP 생산 효율이 급격히 떨어집니다. 포도당 한 분자에서 무산소 조건에서는 2개의 ATP만 생성되지만, 산소가 있으면 약 30에서 32개의 ATP가 생성됩니다. 조직에 산소가 부족하면 에너지 생산이 저해됩니다. 세포 기능이 저하됩니다. 장기간 지속되면 세포가 죽습니다. 저산소는 여러 원인으로 발생합니다. 만성 폐쇄성 폐질환, 폐섬유화, 폐렴 등으로 폐의 가스 교환이 저하되는 폐 문제가 있습니다. 심부전으로 심박출량이 감소하면 조직으로의 산소 전달이 줄어드는 심장 문제가 있습니다. 동맥경화, 혈전 등으로 혈관이 좁아지거나 막히면 해당 조직이 저산소 상태가 되는 혈관 문제가 있습니다. 적혈구나 헤모글로빈이 부족하면 산소 운반 능력이 저하되는 빈혈이 있습니다. 고지대에서는 대기압과 산소 분압이 낮아 저산소 상태가 됩니다.
세포는 산소 농도를 감지하고 반응하는 정교한 시스템을 가지고 있습니다. 그 중심에 HIF, 저산소 유도 인자가 있습니다. 2019년 노벨 생리의학상이 이 시스템의 발견자들에게 수여되었습니다. HIF는 유전자 발현을 조절하는 전사인자입니다. 정상 산소 조건에서 HIF는 빠르게 분해됩니다. 저산소 조건에서는 HIF가 분해되지 않고 축적됩니다. 축적된 HIF는 핵으로 이동하여 수백 개의 유전자 발현을 조절합니다. HIF가 활성화하는 유전자들은 저산소 환경에서 생존을 돕습니다. 적혈구 생성을 촉진하는 에리스로포이에틴, 혈관 형성을 촉진하는 VEGF, 해당과정을 촉진하는 효소들 등입니다.
최근 연구는 저산소 상태에서 분비되는 에리스로포이에틴의 놀라운 이면을 밝혀냈습니다. 에리스로포이에틴은 단순히 적혈구만 만드는 것이 아니라, 파골세포를 자극하여 뼈를 녹입니다. 왜 그럴까요? 적혈구가 만들어지는 장소는 뼈 내부의 골수입니다. 산소가 부족해 적혈구를 급히 많이 만들어야 한다면, 공장인 골수 공간을 확장해야 합니다. 그래서 몸은 뼈 안쪽을 깎아내어 공간을 확보하는 선택을 합니다. 숨이 차면 뼈가 녹습니다. 이것은 비유가 아니라 생리학적 사실입니다.
저산소와 HIF 신호는 뼈 대사에도 영향을 미칩니다. 이 분야의 연구는 아직 진행 중이며, 결과가 복잡합니다. 파골세포에 대한 영향을 보면, 파골세포는 저산소 환경에서 활성화됩니다. 뼈 흡수 구역은 파골세포가 밀봉한 구역으로, 산소 농도가 낮습니다. 저산소가 파골세포 분화와 생존을 촉진한다는 연구들이 있습니다. 골아세포에 대한 영향은 더 복잡합니다. 실제로 골아세포 계열 세포에서 HIF를 과활성화한 동물에서는 골량이 증가했다는 실험도 있고, 반대로 전신적인 심한 저산소나 만성 질환 상황에서는 뼈가 빠르게 소실되는 연구도 있습니다. 국소적이고 단기적인 저산소와 전신적이고 만성적인 저산소의 효과가 다를 수 있습니다. HIF는 VEGF를 통해 혈관 형성을 촉진하는데, 뼈에서 혈관 형성은 골 형성과 밀접하게 연결되어 있습니다. 이 측면에서 HIF는 뼈 건강에 긍정적일 수 있습니다. 전체적으로, 만성 저산소 상태는 뼈 건강에 부정적인 영향을 미치는 것으로 보입니다. 임상 연구들이 이를 지지합니다.
만성 저산소 질환과 뼈 손실의 연결은 여러 질환에서 관찰됩니다. 만성 폐쇄성 폐질환 환자에서 골다공증 유병률이 매우 높아 약 30에서 60퍼센트로 보고됩니다. 저산소, 만성 염증, 글루코코르티코이드 사용, 활동 감소, 영양 불량 등 여러 요인이 기여합니다. 그러나 이 요인들을 보정해도 만성 폐쇄성 폐질환 자체가 골다공증의 독립적 위험인자입니다. 폐쇄성 수면무호흡증에서는 반복적인 저산소가 발생하며, 수면무호흡증과 골다공증의 연관성이 보고되어 있습니다. 수면무호흡증처럼 밤새 숨이 멈췄다 다시 쉬는 간헐적 저산소는 교감신경 활성과 산화 스트레스를 반복적으로 올립니다. 단순한 산소 부족을 넘어 호르몬, 염증, 뼈 대사를 동시에 자극하는 형태의 저산소 스트레스입니다. 심부전 환자에서도 골다공증 유병률이 높습니다. 조직 저관류와 저산소가 기여할 수 있습니다. 만성 빈혈 상태, 특히 겸상적혈구빈혈이나 지중해빈혈에서 뼈 문제가 흔합니다. 골수 과형성으로 인한 뼈 변형, 그리고 골다공증이 발생합니다. 만성 저산소와 높은 골 회전율이 기여합니다. 에리스로포이에틴의 역설이 여기서도 작동합니다. 고지대 거주민에서 골밀도가 낮다는 보고들도 있습니다. 만성 저산소가 기여할 수 있습니다. 그러나 신체 활동, 영양 상태 등 다른 요인들도 있어 해석이 복잡합니다.
진화적 관점에서 저산소는 무엇을 의미할까요? 저산소는 산소 공급에 심각한 문제가 있음을 나타냅니다. 폐가 제대로 기능하지 않거나, 심장이 제대로 펌프질하지 못하거나, 혈액이 산소를 운반하지 못합니다. 어느 쪽이든 개체의 가장 기본적인 생리 기능에 문제가 있습니다. 자연환경에서 만성 저산소 상태의 개체는 오래 생존하지 못했을 것입니다. 활동 능력이 저하됩니다. 포식자를 피하지 못합니다. 먹이를 잡지 못합니다. 번식 성공률이 떨어집니다. 저산소에 대한 반응으로 HIF가 활성화됩니다. HIF는 당장의 생존을 위한 적응을 촉진합니다. 해당과정을 촉진하여 산소 없이도 ATP를 생산합니다. 적혈구 생산을 촉진하여 산소 운반 능력을 높입니다. 혈관 형성을 촉진하여 산소 전달을 개선합니다. 이런 반응들은 단기적 생존에 도움이 됩니다. 그러나 장기적으로는 대가가 있습니다. 해당과정은 효율이 낮고 젖산을 생성합니다. HIF의 만성적 활성화는 염증과 섬유화를 촉진할 수 있습니다. 그리고 뼈 대사에도 영향을 미칩니다. 저산소 상태에서 뼈를 유지하는 것은 자원 낭비일 수 있습니다. 산소가 부족하면 에너지 생산이 제한됩니다. 한정된 에너지를 어디에 쓸 것인가? 당장의 생존인 호흡, 심장 박동, 뇌 기능이 우선입니다. 뼈 유지는 후순위입니다.
DIAH의 상호작용
지금까지 결핍, 염증, 산증, 저산소를 각각 살펴보았습니다. 그러나 실제로 이 트리거들은 독립적으로 작용하지 않습니다. 서로 연결되어 있고, 서로를 강화합니다. 결핍과 염증을 생각해봅시다. 칼슘과 비타민D 결핍은 면역 기능을 저하시킵니다. 비타민D는 면역 조절에 중요한 역할을 합니다.
비타민D 결핍은 감염 감수성을 높이고, 자가면역 질환 위험을 높입니다. 감염과 자가면역은 염증을 유발합니다. 반대로, 만성 염증은 식욕을 저하시키고 흡수를 방해하여 영양 결핍을 악화시킵니다. 염증과 산증을 생각해봅시다. 염증 상태에서 대사가 변합니다. 해당과정이 증가합니다. 젖산이 생성됩니다. 대사성 산증이 유발될 수 있습니다. 반대로, 산증은 염증 반응에 영향을 미칩니다. 산성 환경에서 일부 면역세포 기능이 변합니다.
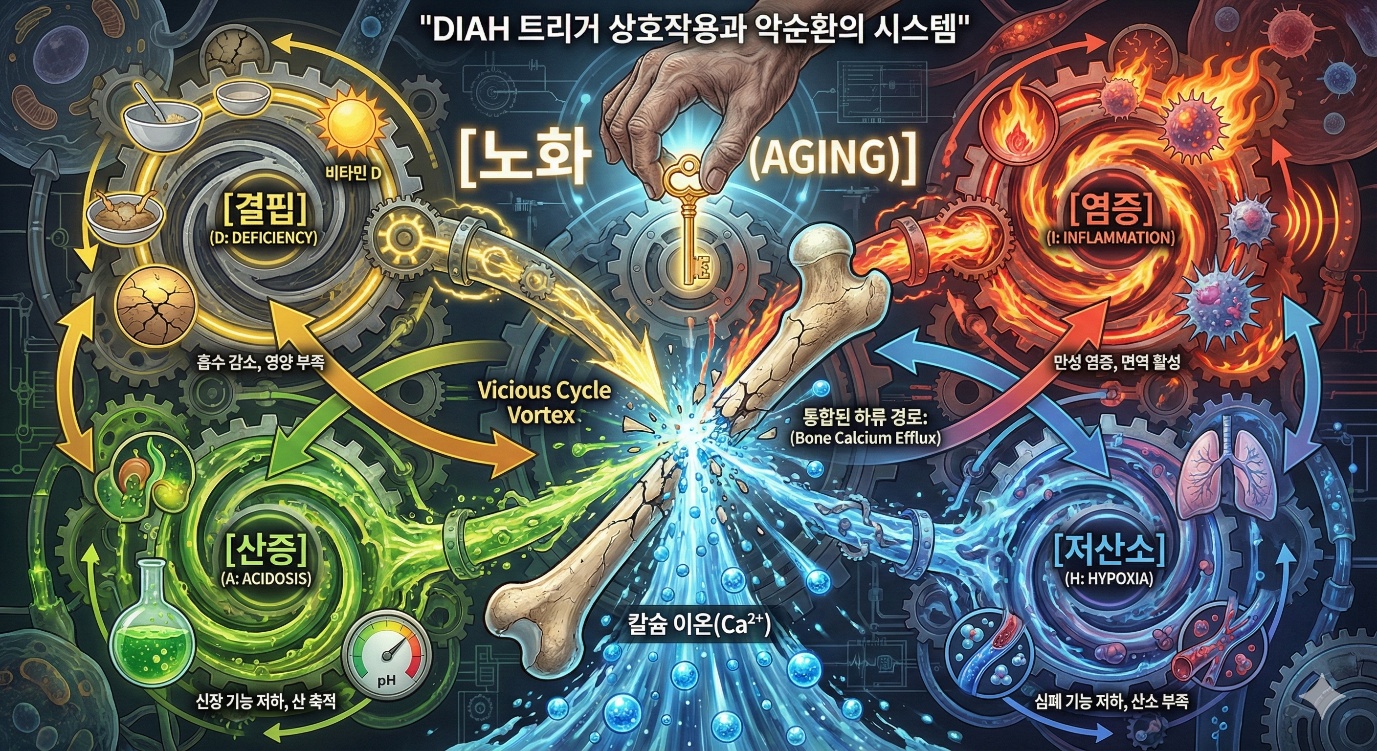
산증과 저산소를 생각해봅시다. 조직 저산소는 무산소 해당을 유발합니다. 젖산이 축적됩니다. 젖산 산증이 발생합니다. 반대로, 산증은 산소 해리 곡선에 영향을 미칩니다. pH가 낮아지면 헤모글로빈의 산소 친화도가 낮아져 조직에 산소가 더 잘 공급됩니다. 이것이 보어 효과입니다. 이것은 보상 기전이지만, 만성 상태에서는 복잡한 상호작용이 있습니다. 결핍과 저산소를 생각해봅시다. 철분 결핍은 빈혈을 유발합니다. 빈혈은 조직 저산소를 유발합니다. 반대로, 만성 폐쇄성 폐질환 같은 만성 저산소 질환에서는 식욕 저하와 흡수 장애로 영양 결핍이 흔합니다. 이렇게 DIAH는 서로 연결되어 있습니다. 하나가 시작되면 다른 것들을 유발하거나 악화시킵니다. 악순환이 형성됩니다. 이 네 가지는 서로의 꼬리를 물고 돌아가는 톱니바퀴와 같습니다.
정리하면, 결핍은 부갑상선호르몬을 통해, 염증은 RANKL/OPG 비율 변화를 통해, 산증은 산성 미세환경과 직접적 무기질 용해를 통해, 저산소는 HIF 신호와 에리스로포이에틴을 통해 모두 파골세포 활성화에서 뼈 칼슘 유출로 이어지는 하나의 하류 경로로 합류합니다. 네 가지 다른 원인이 같은 결과를 낳습니다. 뼈에서 칼슘이 빠져나옵니다. 앞서 제5장에서 언급했듯이, 노화는 DIAH 모든 트리거를 당깁니다. 노화가 칼슘과 비타민D 흡수 감소를 유발하여 결핍으로 이어지고, 노화가 염증노화를 유발하여 염증으로 이어지고, 노화가 신장 기능 저하를 유발하여 산증 경향으로 이어지고, 노화가 심폐 기능 저하를 유발하여 저산소 경향으로 이어집니다. 노화라는 한 단어 안에는 사실 결핍, 염증, 산증, 저산소가 동시에 서서히 켜지는 과정이 숨어 있습니다. 이 네 축이 함께 뼈를 향해 이제 비상 칼슘을 꺼내 쓰라는 신호를 보내는 셈입니다.
이것이 우연일까요? 아니면 설계일까요? 제5장에서 제안했듯이, 이것은 쇠퇴의 열쇠가 작동하는 것으로 해석할 수 있습니다. 번식이 끝나면 보호 메커니즘이 약해집니다. 여러 시스템이 동시에 같은 방향으로 변합니다. DIAH 트리거들이 활성화됩니다. 뼈에서 칼슘이 빠져나옵니다.
쇠퇴가 진행됩니다. 만약 이것이 설계라면, DIAH는 개체의 상태를 평가하는 시스템으로 볼 수 있습니다. 결핍이 있으면 먹이를 구하지 못하고 있습니다. 염증이 있으면 손상이나 감염이 있습니다. 산증이 있으면 대사에 문제가 있습니다. 저산소가 있으면 호흡이나 순환에 문제가 있습니다. 어느 하나라도 있으면 개체가 최적 상태가 아닙니다. 여러 개가 있으면 더욱 그렇습니다. 노화한 개체는 이 모든 트리거가 활성화됩니다. 시스템은 이 개체가 최적 상태가 아니라고 판단합니다. 장기 투자인 뼈 유지에서 자원을 회수합니다.
현대 의학은 DIAH를 개별적으로 치료할 수 있습니다. 결핍은 보충으로 치료합니다. 칼슘 보충제, 비타민D 보충제, 영양 지원. 염증은 항염증 치료로 관리합니다. 비스테로이드성 항염증제, 스테로이드, 생물학적 제제. 산증은 중탄산염 보충이나 원인 치료로 관리합니다. 만성 신장 질환에서 중탄산나트륨을 사용합니다. 저산소는 산소 공급으로 치료합니다. 산소 요법, 원인 질환 치료. 이런 치료들이 개별 트리거를 억제합니다. 그러나 몸의 기본 시스템은 여전히 작동합니다.
트리거 신호가 있으면 뼈에서 칼슘을 동원하는 반응이 일어납니다. 현대 의학의 개입에도 불구하고, 만성질환 환자에서 골다공증이 흔한 이유입니다. 통합적 접근이 필요합니다. 개별 트리거를 치료하는 것과 함께, 뼈 자체를 보호하는 치료도 필요합니다.
결론
이 장에서 우리는 DIAH 칼슘 유출 트리거의 메커니즘을 살펴보았습니다. 결핍은 칼슘과 비타민D의 부족입니다. 외부 공급이 불충분하면 부갑상선호르몬이 뼈에서 칼슘을 동원합니다. 노화에 따른 흡수 감소가 결핍을 유발합니다. 염증은 종양괴사인자 알파, 인터루킨-1, 인터루킨-6 같은 염증성 신호물질을 통해 RANKL/OPG 비율을 높이고 파골세포를 활성화합니다. 면역세포가 직접 RANKL을 분비하기도 합니다. 염증노화가 만성 염증을 유발합니다. 산증은 뼈의 무기질을 완충제로 사용하게 합니다. 물리화학적 용해와 세포 매개 흡수가 모두 증가합니다. 신장 기능 저하와 현대 식이의 산 부하가 기여합니다. 혈액 pH가 정상이어도 뼈는 이미 희생하고 있을 수 있습니다. 저산소는 HIF 경로를 통해 뼈 대사에 영향을 미칩니다. 에리스로포이에틴은 적혈구를 만들기 위해 뼈 안쪽 공간을 확보하려고 파골세포를 자극합니다. 만성 폐질환, 심부전, 빈혈 등에서 뼈 손실이 증가합니다.
이 DIAH 트리거들은 독립적이지 않습니다. 서로 연결되어 악순환을 형성합니다. 노화는 모든 트리거를 동시에 활성화시킵니다. 진화적 관점에서 DIAH는 개체 상태를 평가하는 시스템으로 볼 수 있습니다. 칼슘 유출 트리거가 활성화되면 개체가 최적 상태가 아니라는 신호입니다. 이 신호에 대한 반응으로 뼈에서 칼슘이 방출됩니다. 장기 투자에서 단기 생존으로 자원이 재배분됩니다. 제7장에서는 유출된 칼슘이 어떻게 7M 메커니즘을 통해 만성질환으로 이어지는지 살펴보겠습니다.
참고문헌
1. Harada, S., & Rodan, G. A. (2003). Control of osteoblast function and regulation of bone mass. Nature, 423(6937), 349-355. doi:10.1038/nature01660
2. Boyle, W. J., Simonet, W. S., & Lacey, D. L. (2003). Osteoclast differentiation and activation. Nature, 423(6937), 337-342. doi:10.1038/nature01658
3. Khosla, S. (2001). Minireview: the OPG/RANKL/RANK system. Endocrinology, 142(12), 5050-5055. doi:10.1210/endo.142.12.8536
4. Brown, E. M., & MacLeod, R. J. (2001). Extracellular calcium sensing and extracellular calcium signaling. Physiological Reviews, 81(1), 239-297. doi:10.1152/physrev.2001.81.1.239
5. Holick, M. F. (2007). Vitamin D deficiency. New England Journal of Medicine, 357(3), 266-281. doi:10.1056/NEJMra070553
6. Weitzmann, M. N., & Pacifici, R. (2006). Estrogen deficiency and bone loss: an inflammatory tale. Journal of Clinical Investigation, 116(5), 1186-1194. doi:10.1172/JCI28550
7. Franceschi, C., et al. (2000). Inflamm-aging: an evolutionary perspective on immunosenescence. Annals of the New York Academy of Sciences, 908(1), 244-254. doi:10.1111/j.1749-6632.2000.tb06651.x
8. Kraut, J. A., & Madias, N. E. (2010). Metabolic acidosis: pathophysiology, diagnosis and management. Nature Reviews Nephrology, 6(5), 274-285. doi:10.1038/nrneph.2010.33
9. Bushinsky, D. A. (2001). Acid-base imbalance and the skeleton. European Journal of Nutrition, 40(5), 238-244. doi:10.1007/s394-001-8352-8
10. Arnett, T. R. (2010). Acidosis, hypoxia and bone. Archives of Biochemistry and Biophysics, 503(1), 103-109. doi:10.1016/j.abb.2010.07.021
11. Semenza, G. L. (2012). Hypoxia-inducible factors in physiology and medicine. Cell, 148(3), 399-408. doi:10.1016/j.cell.2012.01.021
12. Rauner, M., et al. (2017). Perspective: hypoxia and bone. BoneKEy Reports, 6, 869. doi:10.1038/bonekey.2016.36
13. Ilich, J. Z., & Kerstetter, J. E. (2000). Nutrition in bone health revisited: a story beyond calcium. Journal of the American College of Nutrition, 19(6), 715-737. doi:10.1080/07315724.2000.10718070
14. Ginaldi, L., et al. (2005). Osteoporosis, inflammation and ageing. Immunity & Ageing, 2(1), 14. doi:10.1186/1742-4933-2-14
15. Lacey, D. L., et al. (1998). Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell, 93(2), 165-176. doi:10.1016/S0092-8674(00)81569-X